Background & Quickstart¶
ortho_seqs¶
Ortho_seqs is a command line tool that implements a mathematical approach to convert sequence data (DNA/protein) to multivariate tensor-valued orthogonal polynomials and project phenotypes onto the polynomial space. We are currently working to update this mathematical approach and will post these updates soon.
We do this by first converting the sequence information into 4-dimensional (for DNA) or 20-dimensional (for amino acids) vectors. The method can also be used for padded sequences to deal with unequal sequence lengths. Find out more about the original approach in this paper Analyzing genomic data using tensor-based orthogonal polynomials with application to synthetic RNAs. The paper gives an example of this method as applied to a case of synthetic RNA from a previously published dataset.
For example, the sample data inputs for this tool are shown in this image. Here, each site in a sequence is first converted to a 4-dimensional vector. The input data includes phenotype values for each sequence.
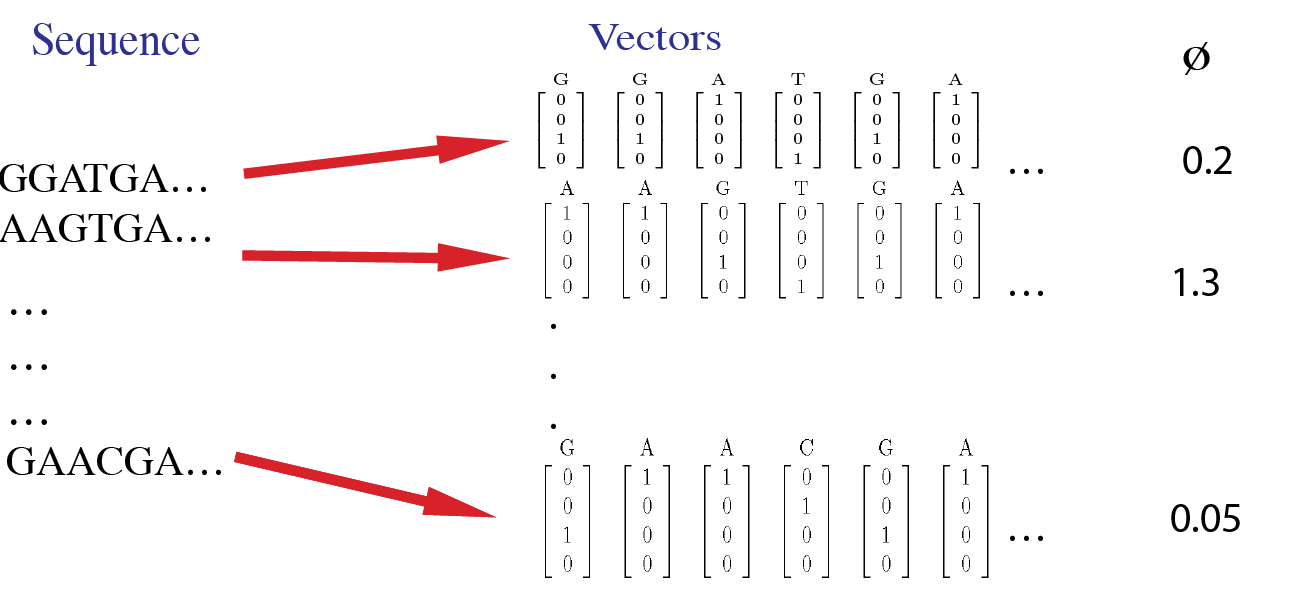
Documentation¶
To view documentation and detailed tutorials for ortho_seqs, click here.
Usage¶
First, install an environment with dependencies for this package:¶
conda create -n ortho_seqs pip
conda activate ortho_seqs
pip install -r requirements.txt
or
conda env create -f conda_environment.yml
conda activate ortho_seq
Then, install the package:¶
python setup.py install
Gather the input file(s) needed.¶
There are three main ways to submit your sequence and phenotype files to ortho_seqs. The first method is to submit them separately, in their own .txt files. Recently, however, an update was added that allows you to submit them both in the same file. For this to apply: 1) The file must be either a .xlsx or a .csv file. 2) The sequences must be in the first column, and the phenotypes must be in the second column. 3) The columns must not have header names.
If you use a single file for the sequence and phenotype, you would submit the file path where you would submit the sequence file path, and do not include the –pheno_file flag. note: the GUI does not support single-file uploads yet.
The phenotypes must be real numbers.
Then, to run the commandline tool:¶
To start with a test example, you can run the sample command below:
ortho_seq orthogonal-polynomial ./ortho_seq_code/tests/data/nucleotide/first_order/test_seqs_2sites_dna.txt --molecule DNA --pheno_file ./ortho_seq_code/tests/data/nucleotide/first_order/trait_test_seqs_2sites_dna.txt --poly_order second --out_dir ../results_ortho_seq_testing/DNA_2sites_test_run/
The above sample command line is building the tensor-valued orthogonal polynomial space based on the sequence data which consists of 12 sequences, each with two sites. Since these are DNA sequences, the vectors are 4-dimensional. These used to be flags for sites, dimensions, and population size, but new functionality will automatically calculate these. Corresponding to each sequence is a phenotype value (a real number) as given in the phenotype file. For DNA, the tool can run first and second order analyses currently. We’ll implement third order in a future version. For amino acids, the current version supports first order analysis and we hope to expand this in the future.
Amino acids/nucleotides that do not appear in any sequence will be removed from the alphabet when the letters are being converted to first order vectors. For example, if the residue ‘R’ (Arginine) never occurs in the sequence dataset, the first order vectors will now have 19 dimensions (instead of 20) and 20 dimensions (instead of 21) if the sequences are padded with ‘n’. This is done to greatly reduce runtime for larger sequence datasets and for longer sequences. When the program will run, it will return this sentence:
Will be computing p sequences with s sites, and each vector will be d-dimensional.
Where p represents the population size (number of rows in sequence file), s represents the number of sites, and d represents the number of amino acids/nucleotides detected in the sequence file (adds on 1 for lowercase n’s). For the above example, the program will return
Will be computing 12 sequences with 2 sites, and each vector will be 4-dimensional.
Along with regressions on each site independent of one another and onto two sites at a time, the above command also computes Fest which is the phenotype estimated by the regressions. This shows that the mathematical calculations are done correctly as we now have an equation that accurately captures our initial data points. This only works here for sequences with 2 sites. If we had more sites, we’d need to do higher order calculations in order to capture all our combinations. Therefore, when running the tool with more sites, as will probably be the case for most users, even just going up to second order gives us useful information about our system. First order tells us the importance of each site (independent of any correlations it might have with another site) and second order tells the importance of pairs of nucleotides independent of other pairs. Please take a look at the paper linked above to learn more about this method.
Flags & Functionality¶
--pheno_file
Input a file with phenotype values corresponding to each sequence in the sequence file. If you have a .xlsx or .csv file, do NOT use this flag (more details above in the **Gather the Input Files Needed** section).
--molecule
Currently, you can provide DNA or protein sequences. Here, you can also provide sequences of unequal lengths, where sequences will be padded with lowercase 'n's until it has reached the length of the longest sequence.
--poly_order
The order of the polynomials that will be constructed. Currently, one can do first and second order for DNA and first order for protein.
--out_dir
Directory where results can be stored.
--precomputed
Let’s say you have a case where you have the same set of sequences but two different corresponding sets of phenotypes. You can build your sequence space and then project the first set of phenotypes onto this space. Then, if you wish to see how the other set of phenotypes maps onto the same sequence space, you can use this flag so that you’re not wasting time and memory to recompute the space. When doing this, be sure to add your results from the first run to the out_dir when rerunning the command with the precomputed flag.
--alphbt_input
Used to group amino acids/nucleotides together, or specify certain amino acids/nucleotides. If you don’t want to group anything, don’t include this flag when running ortho_seqs. For example, putting ASGR for a protein molecule will tell the program to have 6 dimensions: one for each amino acid specified, and one for z, where every unspecified amino acid or nucleotide will be converted to z, and one for n (whenever sequences have unequal lengths, ortho_seqs will pad the shorter sequences with n at the end). You can also comma-separate amino acids/nucleotides to group them. For example, putting AS,GR will make the vectors 4-dimensional, one for AS, one for GR, one for every other amino acid (z), and one for n.
There are also built-in groups:
protein_pnp will group by polar and non-polar amino acids, every other amino acid, and n.
essential groups by essential and non-essential amino acids, every other amino acid, and n. Group 1: Essential - ILVFWHKTM Group 2: Non-Essential - Everything else Group 3: n (Source: https://www.ncbi.nlm.nih.gov/books/NBK557845/)
alberts groups by categories set by Alberts. Group 1: Basic - KRH Group 2: Acidic - DE Group 3: AVLIPFMWGC Group 4: Everything else Group 5: n (Source: https://www.ncbi.nlm.nih.gov/books/NBK21054/)
sigma groups by categories set by Sigma. Group 1: Aliphatic - AILMV Group 2: Aromatic - FYV Group 3: Polar Neutral - NQCST Group 4: Acidic - KRH Group 5: Basic - DE Group 6: Other - G Group 7: Other - P Group 8: n (Source: https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/protein-biology/protein-structural-analysis/amino-acid-reference-chart)
hbond groups by strength of hydrogen bond attractions. Group 1: Can Make Hydrogen Bonds - NQSTDERKYHW Group 2: Can Not Make Hydrogen Bonds - Everything else Group 3: n The first group is able to make hydrogen bonds, whereas the second group is not.
hydrophobicity groups by hydrophobicity. Group 1: Very Hydrophobic - LIFWVM Group 2: Hydrophobic - CYA Group 3: Neutral - TEGSQD Group 4: Hydrophilic - Everything else Group 4: n The first group is very hydrophobic, the second group is slightly hydrophobic, the third group is neutral, and the last group is hydrophilic.
--min_pct
When ortho_seqs is run, a .csv file of covariances will be saved in the specified path. This matrix of covariances is one of the main results of the program (as shown in {sequence_file_name}.npz output below). The csv file will contain the covariance of each nucleotide at each site with another nucleotide at another site (or amino acids at each site). Suppose there are 5 covariance values of 2, 1, 0, 0, -1. For the percentiles, all unique magnitudes will be considered when assigning covariances, which will be 2, 1, and 0. 0 will be the 0th percentile (therefore, assigning 0 to the –min_pct flag will return every covariance), 1 (and -1) will be 33.33…, and 2 will be 66.66… Specifying 50 as –min_pct will only return the row with the covariance of 2, since only 66.6…>50. The min_pct flag is short for minimum percentile, which will remove any covariances from the .csv file that are below the given percentile. The default value is 75.
--pheno_name
Let’s say you know that your phenotype values represent IC50 values. You could then add –pheno_name IC50 as a flag, and on the rFon1D plot that is automatically generated, the y-axis label will include IC50. Default is None.
Results & Outputs¶
Generating logo plot¶
Before even running the tool, we can generate a logo plot to visualize the different nucleic/amino acids in the sequence dataset. This is implemented as a command line function.
Refer to the logo-plot tutorial on the ReadTheDocs for more information on how to generate this.
Running ortho_seqs¶
The tool will provide updates as the run is progressing regarding which parts of the calculations are done being computed. For example, when the mean is computed, it’ll say “computed mean”. All the different elements that it is computing are different parts of building the multivariate tensor-valued orthogonal polynomial space based on the sequence information. To get a general idea of what the calculations mean, please refer to the supplementary methods in the paper linked above. The program will save outputs in npz format. See below for what is stored.
{sequence_file_name}.npz
This will store the calculations that went into constructing the polynomial space. This also includes information about the statics of our sequence space, such as mean, variance and a matrix of covariances. See figures 4 and for ideas on how mean and the matrix of covariances can be visualized. All of these calculations go into building the orthogonal polynomial space based on sequence information and at this point of the program, we have not connected the phenotype (the functional variable) with the sequence information.
{sequence_file_name}_covs_with_F.npz
This will store the covariance of the phenotype (or trait) with the polynomials. This is when we start connecting the phenotype with the sequence space.
{trait_file_name}_Fm.npz
This contains the mean trait value. This is a scalar.
{trait_file_name}_regressions.npz
This set of files contains the main results which includes the following:
rFon1D: This is the regression of the trait onto the first order conditional polynomial orthogonalized within. This tells us the regression of the phenotype onto each site and onto each nucleotide (or amino acid) at that site independent of any correlations that site might have with other sites. For the case of nucleotides, this can be visualized as bar plots as shown in Figure 6 in the paper linked above.
rFon2D: This gives 4 matrices which give the regression of the pheonotype onto (site1)x(site1), (site 1)x(site 2), (site 2)x(site 1) and (site 2)x(site 2), in that order. The second matrix here is the important one and it is the same as rFon12. See description of rFon12.
rFon12: This is the regression of the trait onto pairs of sites for given nucleotides at each site. These are regressions on (site 1)x(site 2) independent of first order associations. Since we’re looking at 2 sites at a time and there’s a possibility of having 4 nucleotides at each site (for the case of DNA), we can visualize this via a 4x4 matrix as shown in Figure 8 in the paper linked above.
cli_output.txt
Everything that prints out on the CLI, when running orthogonal_polynomial, will be saved to this document, in the defined out_dir.
The rf1d class¶
The newest update to ortho_seqs involves adding a new class of objects, called rf1d (short for rFon1D). To run rf1d, use the CLI, and type in rf1d-viz like you would orthogonal_polynomial when running ortho_seqs.
Note: rf1d-viz requires you to have run orthogonal_polynomial beforehand.
--filename
This is the same as the {trait_file_name}_regressions.npz file that is returned from ortho_seqs, as it contains the rFon1D values that are used.
--alphbt_input
Similarly to orthogonal_polynomial, this flag takes in a comma-separated list of the groupings (Note: this list must be comma-separated for the code to work). orthogonal_polynomial will print out the rf1d form of alphabet input in the CLI before any mathematical calculations are made, which will work if you choose to copy/paste it.
--molecule
Identical to how it is in orthogonal_polynomial. It doesn’t matter much what you put here, as this is purely for visual purposes only.
--phenotype
Identical to how it is in orthogonal_polynomial. This is used as the y-axis labeling for the barplot.
--out_dir
The path where you want the visualizations saved, if applicable.
--action
This flag is where you specify which visualization you want.
Options:
barplot - Prints and saves a barplot of the rFon1D values, and saves it, if an out_dir is specified. This is what is called in orthogonal_polynomial at the end.
density - Prints a histogram plot of the rFon1D values, and saves it, if an out_dir is specified.
summary - Prints out the number of sites and dimensions, the alphabet input, the molecule, and calls sort (explained in further detail below). This is called in orthogonal_polynomial automatically, at the very end of the program. This will be saved to the out_dir as summary.txt Note: there won’t be a separate sort.txt file created.
heatmap - Prints a heatmap of the rFon1D values, and saves it, if an out_dir is specified.
boxplot - Prints a boxplot of the rFon1D values, and saves it, if an out_dir is specified.
sort - This will print out the top 10 rFon1D values by magnitude, including the rFon1D value, the site, and the group it belongs to. This will be saved to the out_dir as sort.txt.
To run the GUI¶
A GUI version of the CLI is also available to make it easier for users to utilize the tool. The GUI allows the user to upload the sequence and phenotype information via an upload button, specify the molecule, the polynomial order they wish to run, and provide the path to the directory which contains precomputed sequence space if the user wishes to project a different phenotype onto the same space (i.e., given same sequence data but different corresponding phenotypes). The GUI is in its early form and will include further updates resembling the cli in future versions.

To run the gui, open a terminal and make sure you’re in the ortho_seqs environment just as you would do if you were running the cli (see above). Then type in the following:
ortho_seq gui
This will pull up the gui window and allow you to input the relevant information.
cov_hist_{trait_file_name}.png
This is a histogram of all non-zero covariances. Its bin width is 0.5.
cov_data_frame_{trait_file_name}.csv
This file is a csv file of covariances between every item at every site. This includes the item ID and site for both items in the pair used to calculate the covariance, the covariance value, the covariance magnitude, and an ID for the pair (s1-g2,s3-g4 represents the pairing of an element from the first group in the alphabet at the second site, and an element from the third group at the fourth site). The sites are one-indexed, meaning a value of 3 for the First Site or Second Site column corresponds to site number three along the sequence.
rFon1D_graph_{trait_file_name}.png
This is a bar plot of all nonzero rFon1D values of every item at every site.
Support¶
If you have specific or general questions, feel free to open an issue and we’ll do our best to address them. If you have any comments, suggestions or would like to chat about this method or similar ideas, feel free to reach out via email at saba.nafees314@gmail.com.
Roadmap¶
We hope to implement third order analysis for DNA in the near future. For amino acids, we hope to implement second order analysis. We’ll add visualization ideas soon but if you have any thoughts on this, please feel free to reach out.
Contribution¶
We hope to make the tool run faster as with higher dimenions and higher order analysis of longer sequence data, we can run into memory and time issues. Any thoughts on this or visualization are welcome.